Qiita Philosophy¶
Plugin ideology¶
Qiita can be seen as an analytical pipeline broker that can apply any specific pipeline, tool, or script to any of its stored data. All the analytical pipelines are autonomous, independently developed, and tested, which facilitates the support of current tools and the development of new ones. This principle is supported via virtual environments and artifacts. Virtual environments for each pipeline gives the freedom of adding any pipeline with any software dependencies to Qiita. Artifacts, basically any file in the system, from raw sequence to contingency tables or even data visualizations, permits the system to store any kind of data but also define within each pipelines which commands and parameters can applied to them.
A Study¶
Qiita’s main entity is the idea of a study. A study can have many samples, with many preparations, that have been sequenced several times, Figure 1.
Additionally, study artifacts have 5 different states: sandboxed, awaiting_approval, private, public and archived. A sandboxed artifact has all operational capabilities in the system but is not publicly available, allowing for quick integration with other studies but at the same time keeping it hidden.
Once a user is satisfied with their study and analysis, they can request to upgrade their preparation and all their artifacts status to ‘private’; this confers additional benefits to the project, including permanent space in the repository. During this time, an administrator will validate their study and its status will change to ‘awaiting_approval’; note that users need to request this transition from, please review Making data Public in Qiita and/or send data to EBI-ENA.
At this stage in Qiita the whole preparation in the study (including raw and all processed data) is private. If the user also requests it, the raw data can be deposited for permanent storage to the European Nucleotide Archive (ENA), part of the European Bioinformatics Institute (EBI). Then, when the user is ready, usually when the main manuscript of the study is ready for publication, the user can make for the preparation and all its artifacts to be ‘public’, both in Qiita and the permanent repository, Figure 2. Finally, when new processing algorithms are available, the older BIOM artifacts are ‘archived’, for long term storage.
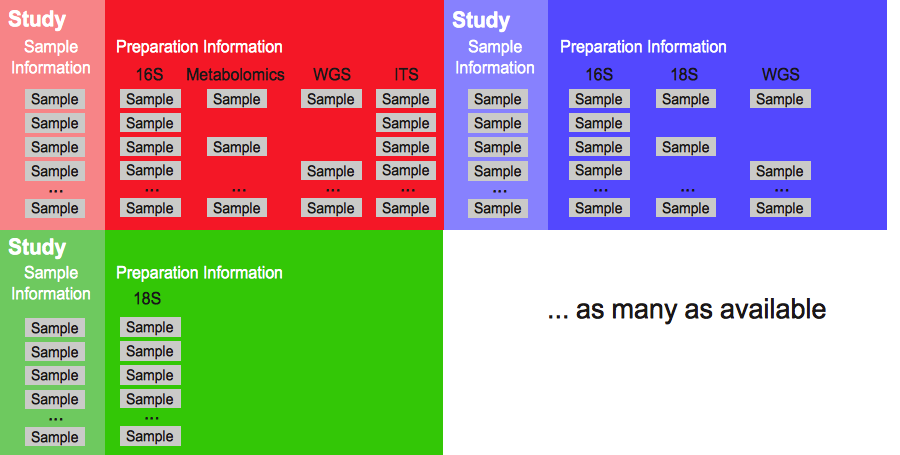
Figure 1. Qiita’s main structure: from single to multiple studies. Increasingly, a simple study is composed of multiple samples which have been prepared using different protocols to identify different microbial features. For example, 16S rRNA amplification to identify the bacteria in or on the sample, metabolomics to identify chemical components formed by the microbial community or within the sample, and/or ITS amplification for identification of fungal organisms that may also be present. Additionally, Qiita allows users to compare their studies with other public ones already available in the system.¶

Figure 2. Possible Qiita artifact states. Artifacts are any file, either uploaded by users or generated by the system. There are 3 possible states: sandboxed, private and public. In the sandboxed and private states no other user has access to the artifacts, unless the owner grants access by sharing the study. In the public state, the artifact is open to all users in the system, and the study can be found by searching from the study listing page.¶
Qiita allows for complex study designs¶
As seen in Figure 1 studies are the main source of data for Qiita, and studies can contain only one set of samples but can also contain multiple sets, each of which can have a different preparations.
The traditional study design includes a single sample and a single preparation information file. However as technology improves, study designs become more complex where a study with a defined set of collected samples can have subsets prepared in different ways so we can answer different questions. For example, let’s imagine a study looking at how different microbial communities changes during mammalian corpse decomposition; thus, your full study design is to collect a set of samples, which you will then process with 16S, 18S and ITS primers. This will result in 1 sample and 3 preparation information files, see it in Qiita.
Now, let’s imagine other more complex example:
All of the samples were prepped for 16S and sequenced in two separate MiSeq runs
50 of the samples were prepped for 18S and ITS, and sequenced in a single MiSeq run
50 of the samples were prepped for WGS and sequenced on a single HiSeq run
30 of the samples have metabolomic profiles
To represent this project in Qiita, you will need to create a single study with a single sample information file that contains all 100 of the samples. Separately, you will need to create four prep information files that describe the preparations for the corresponding samples. All raw data uploaded will need to correspond to a specific preparation (prep) information file. For instance, the data sets described above would require the following data and prep information:
All of the samples prepped for 16S and sequenced in two separate MiSeq runs
1 prep information file describing the two MiSeq runs (use a run_prefix column to differentiate between the two MiSeq runs, more on metadata below) where the 100 samples are represented
the 4-6 fastq raw data files without demultiplexing (i.e., the forward, reverse (optional), and barcodes for each run)
50 of the samples prepped for 18S and ITS, and sequenced in a single MiSeq run
prep information files, one describing the 18S and the other describing the ITS preparations
the 2-3 fastq raw data files (forward, reverse (optional), and barcodes)
50 of the samples prepped for WGS and sequenced on a single HiSeq run
1 prep information files describing how the samples were multiplexed
the 2-3 fastq raw data files (forward, reverse (optional), and barcodes).
NOTE: We currently do not have a processing pipeline for WGS but should soon.
30 of the samples with metabolomic profiles
1 prep information file. the raw data file(s) from the metabolomic characterization.
NOTE: We currently do not have a processing pipeline for metabolomics but should soon.
Portals¶
Qiita allows the hosting of multiple portals within the same infrastructure. This allows each portal to have a subset of studies (often with a similar theme) in a different URL but sharing the same resources. Sharing the same backend resources avoids having multiple sites and data getting out of sync.
The current available portals are:
Main site (all studies from all portals): qiita.microbio.me
Sloan portal (build environment): sloan_microbe.microbio.me
Earth Microbiome Project portal (studies generated under the EMP): emp.microbio.me